-

-
上海角宿企业管理咨询有限公司
Shanghai Spica Management Consulting Co.,Ltd - 17802157742

热门搜索:
上海角宿企业管理咨询有限公司主要提供FDA510(K)、N95认证、TGA注册、欧代注册、自由销售证明、MDR认证,ISO13485体系认证咨询等服务,公司注重规范化内部管理体系建立,在外部对接、内部工作流程、服务后追溯等工作区块,具备良好的策划、评估、监测能力,上海角宿注重企业信用体系建立,全力贯彻商业契约精神。欢迎来电咨询!
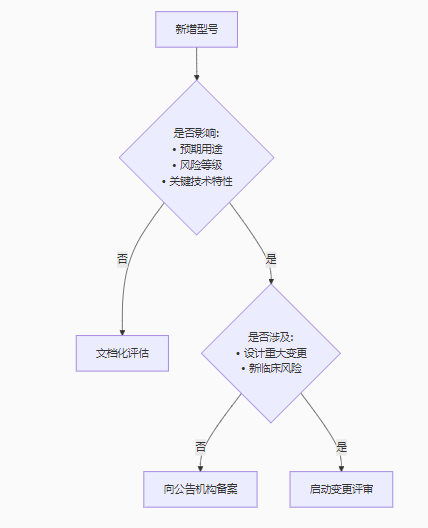
根据欧盟医疗器械法规(MDR 2017/745)*120条,新增型号不一定需要重新申请CE认证,但必须严格评估变更性质。关键判定依据包括:
| 变更类型 | 典型案例 | 合规要求 |
|---|---|---|
| 技术文档更新 (*公告机构介入) | • 外观颜色调整 • 非关键部件供应商变更 | 更新技术文件, 内部评审记录 |
| 重大变更 (需公告机构审查) | • 新增适应症 • 核心算法修改 • 灭菌方式改变 | 提交变更申请, 可能需补充临床评估 |
| 全新产品 (需重新认证) | • 工作原理改变 • 风险等级提升 | 全新MDR认证流程 |
非灭菌/非测量类:可自主更新技术文件
灭菌/测量类:需公告机构审核关键变更
材料变更、软件重大升级需公告机构批准
典型审核周期:4-8周
任何可能影响临床性能的变更均需:
补充临床数据
*小组咨询(如适用)
平均处理时间:3-6个月
产品分类变化(如IIa→IIb)
新增预期医疗用途(如诊断→**)
核心技术创新(如新增AI功能)
同系列扩展:通过MEDDEV 2.1/1 Rev.4评估
参数调整:提供等效性验证报告
配件兼容:更新技术文件附录
针对型号扩展需求,我们提供全流程合规解决方案:
✅ 变更影响评估:72小时出具法律意见书
✅ 技术文件升级:符合MDR Annex II/III要求
✅ 公告机构沟通:加速变更评审流程(合作NB包括TÜV SÜD、BSI等)
✅ 临床评估补充:按MEDDEV 2.7/1 Rev.4执行
✅ 上市后监督:更新PMS/PMCF计划
2024年成功案例:
协助某骨科企业3周内完成:
5个新型号植入物的同系列认证扩展
节省60%认证成本
建立自动化变更管控系统
欧盟MDR监管日趋严格,专业支持决定市场准入效率!
立即联系角宿MDR*团队,获取定制化变更方案。