-

-
上海角宿企业管理咨询有限公司
Shanghai Spica Management Consulting Co.,Ltd - 17802157742

热门搜索:
上海角宿企业管理咨询有限公司主要提供FDA510(K)、N95认证、TGA注册、欧代注册、自由销售证明、MDR认证,ISO13485体系认证咨询等服务,公司注重规范化内部管理体系建立,在外部对接、内部工作流程、服务后追溯等工作区块,具备良好的策划、评估、监测能力,上海角宿注重企业信用体系建立,全力贯彻商业契约精神。欢迎来电咨询!
如何编制符合MDR要求的标签?这是每一个医疗器械制造商都需要面对的问题。根据欧盟医疗器械法规(MDR)的定义,“标签”是指出现在医疗器械本身、每个单元的包装或多个器械的包装上的任何书面、印刷或图形信息。标签的目的是识别医疗器械及其制造商,并传达有关安全性、使用和性能的基本信息,以满足医疗器械用户的需求。
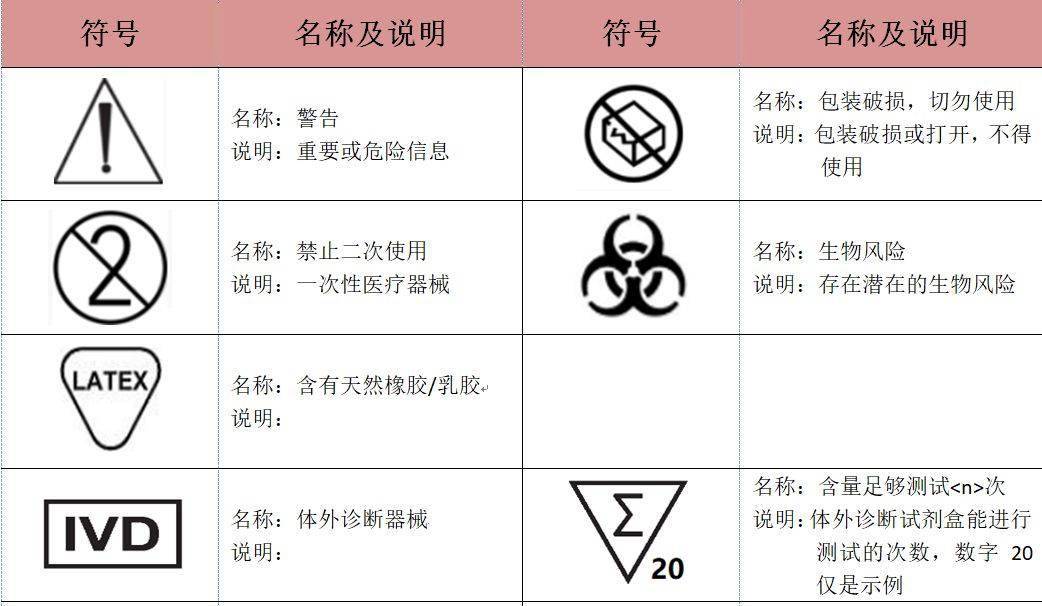
在新的MDR下,医疗器械的标签需要更多的信息,因为器械安全性和临床有效性数据需要与用户(医务人员和患者/最终用户)透明共享。欧盟MDR附录I*三章“一般安全和性能要求”中涵盖了有关医疗器械所提供信息的所有要求。因此,在尝试遵守欧盟MDR标签要求时,需要确保涵盖所有必要的符号和信息,并考虑标签的大小问题。最大的挑战是如何将更多的符号和数据全部贴在标签上。
在标签设计过程中,需要注意以下几点:标签和说明的媒介、格式内容、易读性和位置必须与目标用户的技术知识、经验、教育或培训相配。此外,使用说明必须以目标用户易于理解的术语编写,并在适当的情况下辅以图纸和图表。
标签上的新元素包括UDI编号、警告和注意事项、使用日期或有效期、可重复使用的医疗器械、电子使用说明、用于无菌医疗器械的描述、制造商信息和欧盟授权代表信息以及其他信息。特别需要注意的是,每个医疗器械都必须有标明它是医疗器械,如果设备仅用于临床研究,则标签必须包含“专门用于临床研究”的字样。
准备MDR标签的最佳方法是阅读附件I一般安全和性能要求,*三章,要求23.2和23.3,并找出哪些要求适用于您的医疗器械。定义后,您需要使用ISO 15223-1:2020中的符号创建标签设计。
另外,标签的语言也需要考虑。根据欧盟的要求,需要提供所有欧盟24个成员国官方语言的版本。如果只提供英文版的标签,可能会被开出NG项。因此,制造商需要提供一个程序文件,证明提交的英文版标签是标准参考版,一旦被认可,就会按照这个标准的版本进行多国语言的翻译。