510(K)第三方审核计划作用、流程及适用器械范围
2024-04-24 浏览次数:67次
510(K)第三方审核计划是一种自愿的医疗器械替代审核流程,旨在加快中低风险器械510(k)审核速度,让FDA可以将资源集中高风险器械的审核上。
一、510(K)第三方审核哪些流程
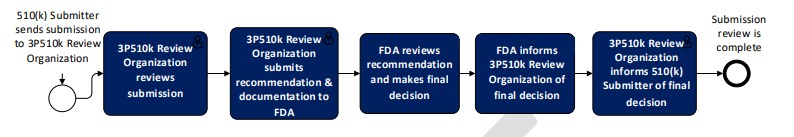
FDA第三方510(k)审核基本流程:510(k)申请人首先向510(k)第三方审核机构递交技术文件,第三方审核机构按照与FDA相同的标准审核完成后向FDA递交推荐意见,FDA审核之后做出最终决定并通知第三方审核机构,第三方审核机构将最终结果通知申请人。
二、哪些器械可申请510(K)第三方审核
目前,符合510(K)第三方审核路径审核要求的器械主要包括 I 类及部分 II 类器械,高风险III类及较为复杂的II类器械不在510(k)第三方审核机构的审核范围内。FDA会动态更新产品列表,使FDA始终可将审查资源集中于高风险及复杂器械,同时保持对第三方审核项目的高度信任。
在向510(K)第三方审核机构提出申请之前,制造商应先浏览FDA官网上的第三方审核的器械清单及不同第三方审核机构所能审核的器械目录。这些清单和目录可以帮助制造商确定自己的产品是否符合第三方审核的要求,以及选择合适的审核机构。
三、哪些机构510(K)第三方审核资质
根据《联邦食品、药品和化妆品法》*523条规定,“当经认可的第三方实质上不符合相关规定时,FDA可以在提供通知后暂停或撤销对任何第三方审查组织的认可。”因此,FDA对第三方审核机构的审核资质要求非常严格。
目前,FDA认可的510(K)第三方审核机构主要包括UL Verification Services Inc.、BSI Group、TÜV SÜD America Inc.等。这些机构均具备丰富的医疗器械审核经验和专业知识,可以为制造商提供高质量的审核服务。
bys0613.b2b168.com/m/